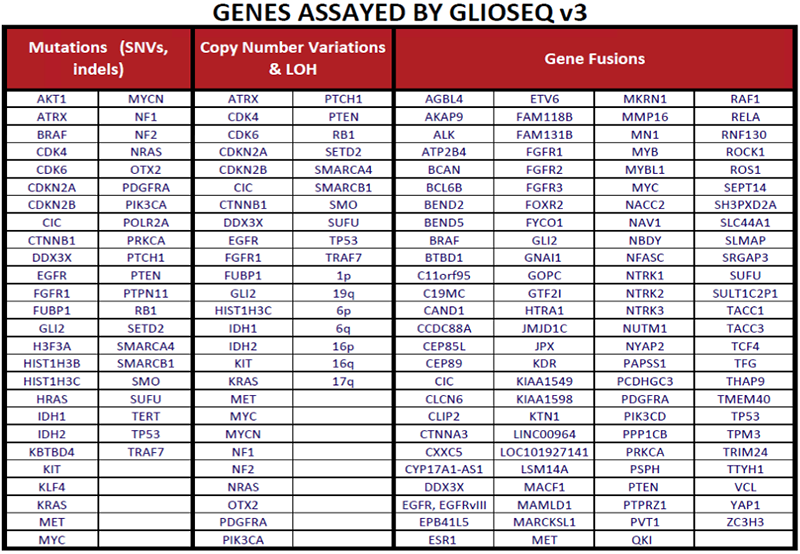
Molecular markers are used to support and enhance the diagnosis, prognosis and treatment of adult and pediatric CNS tumors.
The GlioSeq test identifies genetic alterations that are relevant to different CNS tumors subtypes and grades. [1, 2]
Low grade gliomas (WHO Grade I-II), including pilocytic astrocytoma, pilomyxoid astrocytoma, ganglioglioma,
pleomorphic xanthoastrocytoma, and other neuronal and neuroglial tumors often harbor mutations or gene fusions in BRAF,
PDGFRA,
PTPN11, FGFR family genes, NTRK2, RAF1, and ALK.[3-5] Neurofibromatosis type 1 associated
pilocytic astrocytoma characteristically
harbor mutations and/or loss in NF1 gene, resulting in bi-allelic inactivation of the gene and frequently seen in the younger age
group with multiple tumors located retro-orbitally, along the optic tract, and base of the brain.[6] In addition, genetic
alterations in NTRK3, NTRK1, MYBL1, MYB, FGFR1, and RAF1 genes are commonly
seen in pediatric low-grade gliomas.[2, 3, 7, 8]
Diffusely infiltrative Gliomas (WHO grade II-III) were classified into three glioma subtypes based on
histopathologic features, molecular alterations, and clinical behavior.[9] Oligodendrogliomas harbor
mutations in IDH1 and IDH2 genes, 1p/19q co-deletion, TERT promoter mutation and alterations in CIC,
and FUBP1 genes. Infiltrating astrocytoma harbor IDH1/2, TP53, and ATRX mutations. Both can progress to a
higher-grade glioma by acquiring additional genomic alterations in the RTK-RAS-PI3K pathway genes. A
subset of lower grade gliomas do not harbor IDH mutations but have genetic alterations similar to high
grade gliomas (WHO grade IV) and considered to be a precursor to IDH-mutant (secondary) GBMs.[9-11]
Primary GBMs (WHO grade IV) are IDH wild-type and harbor a number of genetic alterations that lead to
dysregulation of critical signaling pathways including i) receptor tyrosine kinase (RTK)/RAS/PI(3)K pathway via genetic alterations in
EGFR, FGFR3, NTRK1, ROS1, PDGFRA, PIK3CA, RAS, MET,
KIT, PTPN11, NF1, and PTEN genes ii) Cell cycle pathways via inactivating mutation/loss of
TP53, CDKN2A, and RB1 genes and gain of function of CDK4, and CDK6 genes and
iii) TERT promoter mutations.[10] Pediatric high-grade gliomas are unique, featuring mutations and/or copy number alterations in
H3F3A, HIST1H3B, HIST1H3C, SETD2, ATRX, NF1, MYCN, PDGFRA,
and BRAF genes. IDH mutations are rare and usually restricted to adolescent patients.
These tumors can also harbor fusions involving BRAF, FGFR, NTRK1, NTRK2, NTRK3, and
ALK genes [12-16].
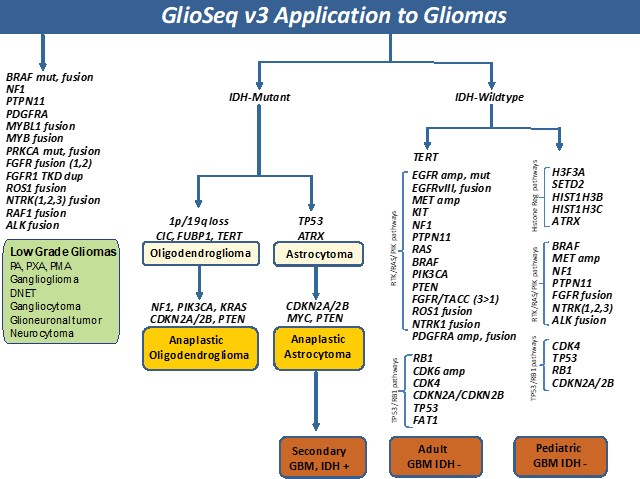
Ependymomas have been classified into molecular subgroups using a combination of genomic alterations and anatomic location along the
neuraxis. Based on presence of mutations and copy number alterations, GlioSeq v3 test is able to established molecular classification
in 76% of ependymomas. [17] RELA gene fusion defines a distinct clinicopathologic subtype of supratentorial ependymomas
associated with chromothripsis, multiple copy number abnormalities, aggressive biologic behavior, and shorter overall survival.
In contrast, supratentorial ependymomas with YAP1 gene fusion, NF2-mutated and 6q-deleted spinal ependymomas as associated
with favorable clinical outcomes.[18]
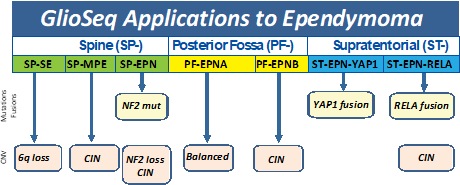
Medulloblastomas have been recently classified into four groups (WNT (wingless), SHH (sonic hedgehog), Group 3, and Group 4) based
on molecular profiling and clinical outcome. Mutations in CTNNB1 and DDX3X, with or without inactivating TP53 alterations, are
typically seen in Wnt-pathway medulloblastomas that are associated with a better prognosis. Tumors with PTCH1, SMO, SUFU, and TERT
promoter alterations and fusions in GLI2, TCF4, and DDX3X genes characterize the SHH class medulloblastomas, and have an intermediate
prognosis between Wnt and group 3/4 tumors. In contrast, mutations in KBTBD4 and OTX2, MYC amplification and fusions involving PVT1,
PIK3CD, and PTEN have been described in group 3 medulloblastomas and MYCN/CDK6 amplification, mutations in KBTBD4 and OTX2, and TP53
rearrangements are more common in the group 4 medulloblastomas. The group 3 and 4 medulloblastomas are far more likely to have a poor
prognosis even with therapy.[19, 20] Other types of CNS embryonal tumors, including atypical teratoid/rhabdoid tumor (ATRT) can
harbor gene rearrangements in C19MC (microRNA cluster region), BEND2, MN1, CIC, and FOXR2 and mutations in SMARCA4 and SMARCB1.[21-23]
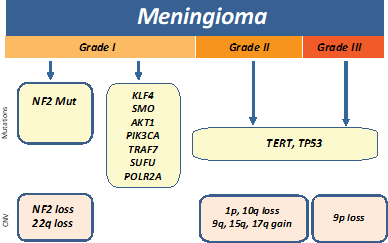
Meningiomas are classified by the WHO into benign meningiomas (grade I), atypical (grade II), and anaplastic (grade III).
Inactivation of NF2 via mutation or loss of 22q is the most common early genetic alteration in meningiomas and found in ~60% of
sporadic cases. Recurrent mutations in KLF4, AKT1, SMO, PIK3CA, TRAF7, SUFU, and
POL2RA genes are often present in NF2-negative
sporadic meningiomas.[24-26] GlioSeq v3 test allows for risk stratification by detecting genetic alterations in genes which are
most often associated with benign tumors (such as TRAF7, AKT1, KLF4, SMO, PIK3CA and POLR2A) and the assessment of alterations
which are associated with malignant progression such as TERT-promoter mutation and multiple copy number alterations including
1p, 10q, and 9p (CDKN2A) loss.
References
- Nikiforova, M.N., et al., Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol, 2016. 18(3): p. 379-87.
- Roy, S., et al., Clinical Utility of GlioSeq Next-Generation Sequencing Test in Pediatric and Young Adult Patients With Brain Tumors. J Neuropathol Exp Neurol, 2019.
- Collins, V.P., D.T. Jones, and C. Giannini, Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol, 2015. 129(6): p. 775-88.
- Picca, A., G. Berzero, and M. Sanson, Current therapeutic approaches to diffuse grade II and III gliomas. Ther Adv Neurol Disord, 2018. 11: p. 1756285617752039.
- Johnson, A., et al., Comprehensive Genomic Profiling of 282 Pediatric Low- and High-Grade Gliomas Reveals Genomic Drivers, Tumor Mutational Burden, and Hypermutation Signatures. Oncologist, 2017. 22(12): p. 1478-1490.
- Gutmann, D.H., et al., Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res, 2013. 23(3): p. 431-9.
- Ellison, D.W., et al., cIMPACT-NOW update 4: diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol, 2019. 137(4): p. 683-687.
- Ramkissoon, L.A., et al., Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A, 2013. 110(20): p. 8188-93.
- Cancer Genome Atlas Research, N., et al., Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med, 2015. 372(26): p. 2481-98.
- Aldape, K., et al., Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol, 2015. 129(6): p. 829-48.
- Brat, D.J., et al., cIMPACT-NOW update 3: recommended diagnostic criteria for "Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV". Acta Neuropathol, 2018. 136(5): p. 805-810.
- Korshunov, A., et al., Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol, 2015. (5): p. 669-78.
- Gajjar, A., et al., Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol, 2015. 33(27): p. 2986-98.
- Korshunov, A., et al., H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol, 2017. 134(3): p. 507-516.
- Wu, G., et al., The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet, 2014. 46(5): p. 444-450.
- Castel, D., et al., Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol, 2015. 130(6): p. 815-27.
- Roy, S., et al., N.e., Utility of GlioSeq next-generation sequencing for molecular classification of ependymomas. Modern Pathology, 2018.
- Pajtler, K.W., et al., Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell, 2015. 27(5): p. 728-43.
- Gajjar, A.J. and G.W. Robinson, Medulloblastoma-translating discoveries from the bench to the bedside. Nat Rev Clin Oncol, 2014. 11(12): p. 714-22.
- Northcott, P.A., et al., The whole-genome landscape of medulloblastoma subtypes. Nature, 2017. 547(7663): p. 311-317.
- Kleinman, C.L., et al., Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet, 2014. 46(1): p. 39-44.
- Sturm, D., et al., New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell, 2016. 164(5): p. 1060-1072.
- Fruhwald, M.C., et al., Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol, 2016. 18(6): p. 764-78.
- Clark, V.E., et al., Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science, 2013. 339(6123): p. 1077-80.
- Yuzawa, S., H. Nishihara, and S. Tanaka, Genetic landscape of meningioma. Brain Tumor Pathol, 2016. 33(4): p. 237-247.
- Clark, V.E., et al., Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet, 2016. 48(10): p. 1253-9.